


Molecular dynamics (MD) simulations model atomic interactions over time and require significant computational power. However, many simulations have small (<400 K atoms) system sizes that underutilize modern GPUs, leaving some compute capacity idle. To maximize GPU utilization and improve throughput, running multiple simulations concurrently on the same GPU using the NVIDIA Multi-Process Service (MPS) can be an effective solution.
This post explains the background of MPS and how to enable it as well as benchmarks of the throughput increase. It also offers some common usage scenarios using OpenMM, a popular MD engine and framework, as an example.
What is MPS?
MPS is an alternative, binary-compatible implementation of the CUDA Application Programming Interface (API). It allows multiple processes to share a GPU more efficiently by reducing context-switching overhead, improving overall utilization. By having all processes share a single set of scheduling resources, MPS eliminates the need to swap scheduling resources on and off the GPU when switching contexts.
Starting with the NVIDIA Volta GPU generation, MPS also allows kernels from different processes to run concurrently, which is helpful when individual processes cannot fully saturate the full GPU. It is supported on all NVIDIA GPUs, Volta generation and later.
One key benefit of MPS is that it can be launched with regular user privilege. Use the following code for the simplest way to enable and disable MPS:
nvidia-cuda-mps-control -d # Enables MPSecho quit | nvidia-cuda-mps-control # Disables MPS |
To run multiple MD simulations concurrently with MPS, launch multiple instances of sim.py, each as a separate process. If you have multiple GPUs on a system, you can use CUDA_VISIBLE_DEVICES to further control the target GPU or GPUs for a process.
CUDA_VISIBLE_DEVICES=0 python sim1.py & CUDA_VISIBLE_DEVICES=0 python sim2.py & ... |
Note that when MPS is enabled, each simulation may become slower, but because you can run multiple simulations in parallel, the overall throughput will be higher.
Tutorial for OpenMM
For this short tutorial, we use OpenMM 8.2.0, CUDA 12, and Python 3.12.
Test configuration
To create this environment, refer to the OpenMM Installation Guide.
conda create -n openmm8.2conda activate openmm8.2conda install -c conda-forge openmm cuda-version=12 |
After installation, test it with the following command:
python -m openmm.testInstallation |
We used the benchmarking script from the openmm/examples/benchmark.py GitHub repo. We ran multiple simulations at the same time with the following code snippet:
NSIMS=2 # or 1, 4, 8for i in `seq 1 NSIMS`;do python benchmark.py --platform=CUDA --test=pme --seconds=60 &done# test systems: pme (DHFR, 23k atoms), apoa1pme (92k), cellulose (409k) |
Benchmarking MPS
Generally speaking, the smaller the system size, the more uplift you can expect. Figures 1, 2, and 3 show the results of applying MPS on three benchmark systems: DHFR (23K atoms), ApoA1 (92K atoms), and Cellulose (408K atoms) using a range of GPUs. The charts show the number of simulations running concurrently on the same GPU and the resulting total throughput.
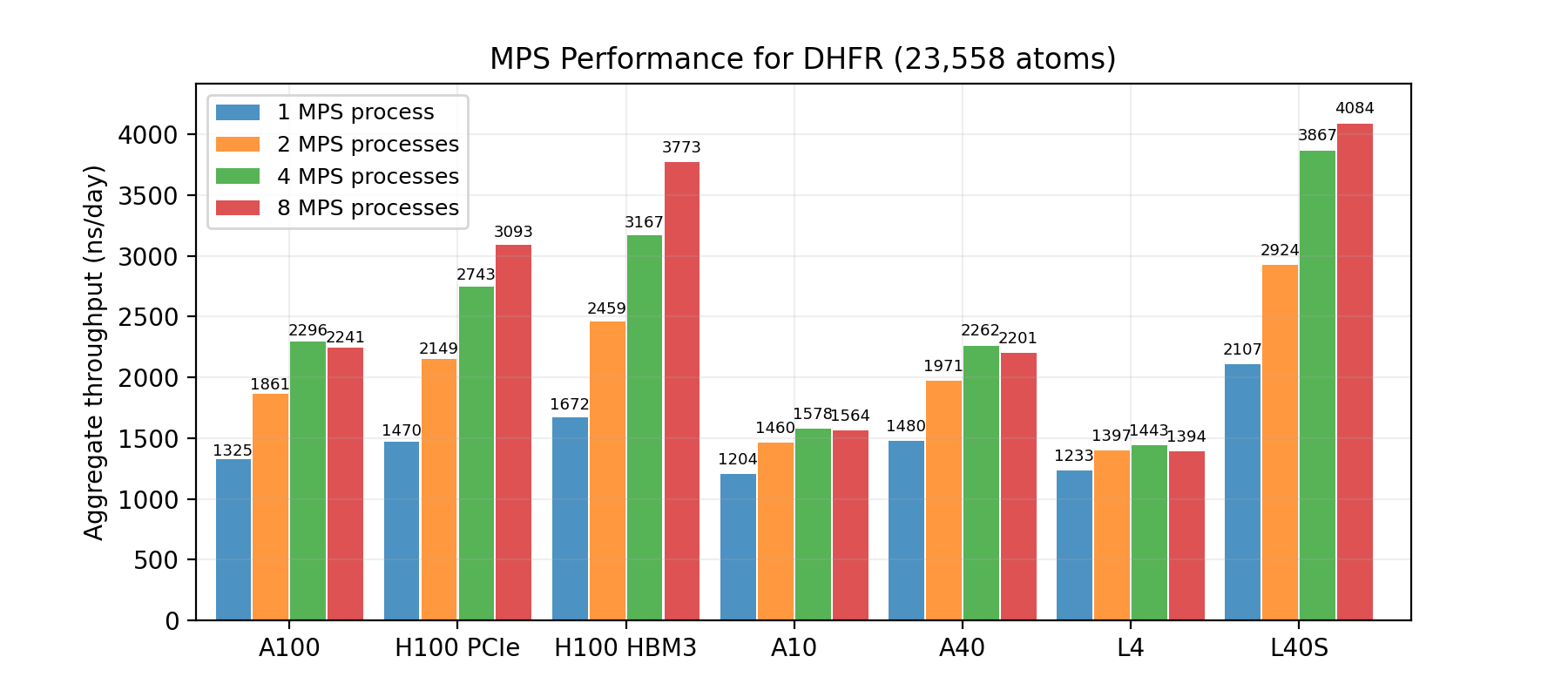
The DHFR test system is the smallest of the three, and therefore enjoys the largest performance uplift with MPS. For some GPUs, including the NVIDIA H100 Tensor Core, the total throughput can more than double.


Even when the system size grows to 409K atoms, as in the Cellulose benchmark, MPS still enables high-end GPUs to achieve about 20% more total throughput.
More throughput with CUDA_MPS_ACTIVE_THREAD_PERCENTAGE
By default, MPS enables all processes to access all resources of a GPU. This can be beneficial for performance, as simulations can take full advantage of all available resources when the other simulations are idle. The force calculation phase of an MD simulation has much more parallelism than the position update phase, allowing for simulations to use more than their fair share of GPU resources improves performance.
However, because multiple processes run simultaneously, it is often unnecessary and can result in destructive interference of performance. CUDA_MPS_ACTIVE_THREAD_PERCENTAGE is an environment variable that enables users to set the maximum percentage of threads available to a single process, and this can be used to increase throughput further.
To achieve this result, modify the code snippet:
NSIMS=2 # or 1, 4, 8export CUDA_MPS_ACTIVE_THREAD_PERCENTAGE=$(( 200 / NSIMS )) for i in `seq 1 NSIMS`;do python benchmark.py --platform=CUDA --test=pme --seconds=60 &done |

CUDA_MPS_ACTIVE_THREAD_PERCENTAGE further increases throughput for some GPUsThis testing shows that a percentage of 200 / number of MPS processes results in the highest throughput. With this setting, the collective throughput for 8 DHFR simulations further increases by about 15 – 25% and approaches 5 microseconds per day on a single NVIDIA L40S or NVIDIA H100 GPU. This is more than doubling the throughput from a single simulation on a GPU.
MPS for OpenFE free energy calculations
Estimating free energy perturbation (FEP) is a popular application of MD simulations. FEP relies on replica-exchange molecular dynamic (REMD) simulations, where multiple simulations at different λ windows run in parallel and exchange configurations to enhance sampling. Within the OpenMM ecosystem, the OpenFreeEnergy (OpenFE) package provides protocols based on the multistate implementations in openmmtools. However, those simulations are run with OpenMM context switching, where only one simulation is conducted at a time. As a result, it suffers from the same issue of GPU underutilization.
MPS can be used to address this issue. With OpenFE installed, running an FEP leg is done with the following command:
openfe quickrun <input> <output directory> |
Following the same logic, you can run multiple legs at the same time:
nvidia-cuda-mps-control -d # Enables MPSopenfe quickrun <input1> <output directory> &openfe quickrun <input2> <output directory> ... |
The time to run the equilibration phase of replica exchange simulations is measured, which includes 12 * 100 ps simulations. The simulations are run on an L40S or an H100 GPU. We observed 36% higher throughput using three MPS processes.

Get started with MPS
MPS is an easy-to-use tool that increases MD simulation throughput without much coding effort. This post explored the uplift in throughput on different GPUs with several different benchmarking systems. We looked at further increasing throughput using CUDA_MPS_ACTIVE_THREAD_PERCENTAGE. We also applied MPS to OpenFE free energy simulations and observed an increase in throughput.
Learn more about MPS in the on-demand NVIDIA GTC session, Optimizing GPU Utilization: Understanding MIG and MPS. You can ask your MPS implementation questions on the NVIDIA Developer CUDA forum.
To get started with OpenMM molecular dynamics simulation code, check out the OpenMM tutorials.
Maximizing OpenMM Molecular Dynamics Throughput with NVIDIA Multi-Process Service